Protein structure prediction
Genome-wide sequencing projects generate extensive data on genes, transcripts, and proteins. However, experimental methods for determining protein tertiary and quaternary structures face significant technical challenges and are not keeping pace with the data influx. This discrepancy underscores the need for predictive methods that offer reliable macromolecular models and insights into protein structures without experimental resolution. Simplifying prediction involves representing 3D structures as sequences of structural assignments, recasting the task as a classification problem. For instance, assigning a secondary structure state or solvent accessibility value to each residue results in one-dimensional strings (Figure 1), providing initial insights into protein structure and function.
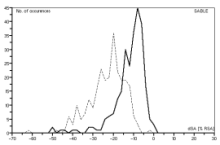
To enhance protein structure prediction and functional annotation, accurate prediction of amino acid residues' relative solvent accessibilities (RSAs) is crucial. Our novel method, SABLE, significantly improves RSA prediction (Ref 1). Unlike other machine learning approaches that typically enforce arbitrary class boundaries, SABLE employs non-linear regression with feedforward and recurrent neural networks for a continuous approximation of RSA. This consensus predictor approach differs from traditional classification methods.
Validation on a diverse non-redundant protein set demonstrates SABLE's effectiveness in enhancing 1D prediction methods for various protein structural characteristics. Incorporating predicted RSA into models reduces feature count and improves generalization. Notably, RSA prediction enhances accuracy in secondary structure (Ref 2), trans-membrane domain (Ref 3), disorder region, and phosphorylation site prediction (Ref 4).
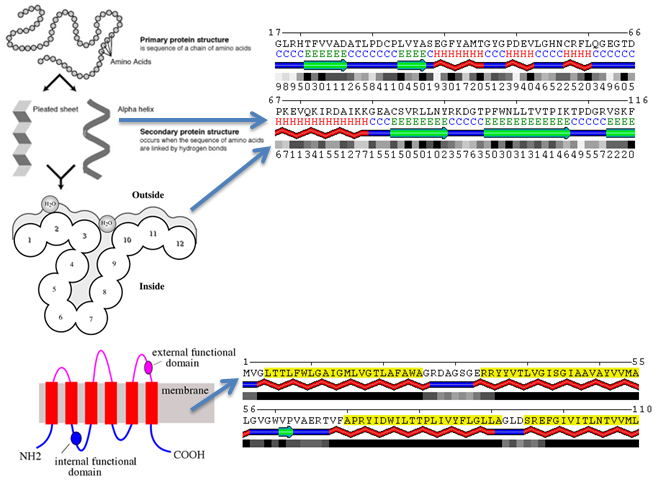
Figure 1.
Schematic Representation of Protein Structural Features as Character Strings. Specifically, α-helices are denoted by 'H', β-sheets by 'E', and unstructured loops by 'C'. The degree of solvent exposure is quantified by relative solvent accessibility, expressed as a percentile. Transmembrane regions and soluble segments are coded as 'T' and 'N', respectively, while here transmembrane regions are accentuated in yellow.
References
- Adamczak R, Porollo A, Meller J. Accurate prediction of solvent accessibility using neural networks-based regression. Proteins. 2004 Sep 1;56(4):753-67. PubMed PMID: 15281128.
- Adamczak R, Porollo A, Meller J. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins. 2005 May 15;59(3):467-75. PubMed PMID: 15768403.
- Cao B, Porollo A, Adamczak R, Jarrell M, Meller J. Enhanced recognition of protein transmembrane domains with prediction-based structural profiles. Bioinformatics. 2006 Feb 1;22(3):303-9. Epub 2005 Nov 17. PubMed PMID: 16293670.
- Swaminathan K, Adamczak R, Porollo A, Meller J. Enhanced prediction of conformational flexibility and phosphorylation in proteins. Adv Exp Med Biol. 2010;680:307-19. PubMed PMID: 20865514.